
Actualizado 18 mayo 2023
Definición
Enfermedad autoinmune ampollosa subepidérmica más frecuente.
Fisiopatología
Respuesta inmune desregulada de las células T y síntesis de autoanticuerpos IgG contra las proteínas hemidesmosomas BP180 (BPAg2 o colágeno tipo XVII) y BP230 (BPAg1), componentes de la unión dermoepidérmica.
IgG1 e IgG3 predominan entre las subclases circulantes de IgG activadoras de la enfermedad. También involucra IgE y mastocitos.
Esta unión promueve la activación y depósito del complemento, el reclutamiento de células inflamatorias, la quimiotaxis de neutrófilos, la liberación de enzimas proteolíticas, la activación de la cascada inflamatoria y la degradación con separación de la zona de la membrana basal.
Presentación clínica
- Responsable del 80% de las dermatosis ampollosas subepidérmicas.
- Afecta principalmente a ancianos > 70-80 años y casos raros en niños y adolescentes.
- No tiene predilección por el sexo, pero algunos dicen que afecta más a las mujeres hasta los 75 años y luego afecta a más hombres.
- El aumento de la incidencia en los últimos años se atribuyó al envejecimiento de la población con múltiples comorbilidades y exposición a fármacos que pueden desencadenar la enfermedad, además de un aumento en el diagnóstico de presentación no ampollosa y en la precisión de las técnicas de laboratorio.
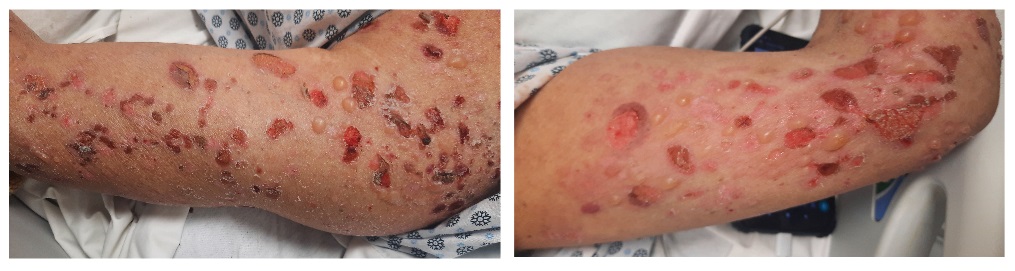
Cuadro clínico:
- Cuadro clásico:
- Ampollas tensas, hialinas o hemorrágicas, de 1-4 cm, sobre placa eritematosa urticariforme o piel sana.
- Localizados en tronco y extremidades, principalmente axilas, abdomen inferior, regiones inguinales y cara interna de muslos.
- La afectación suele ser simétrica y deja una costra y erosión en el sitio.
- Puede tener una fase eritematosa urticariforme no ampollosa prodrómica semanas o meses antes de las ampollas.
- Puede ser localizada (más infrecuente) o diseminada.
- Acompañado de prurito.
- Afectación de la mucosa oral en el 10-30% de los casos. Más raramente puede afectar la mucosa ocular, nasal, faríngea, esofágica y genital.
- Puede dejar hiperpigmentación residual y milia.
- Penfigoide ampolloso no ampolloso:
- Placas urticariformes, pruriginosas, que pueden persistir durante semanas o meses en el mismo lugar que el cuadro clásico.
- Puede haber escoriaciones, pápulas y nódulos.
- Puede haber eritema multiforme similar o prurigo similar.
- Puede representar la fase prodrómica (9.8% de los casos) u otra variante clínica.
- Penfigoide ampolloso en niños y adolescentes:
- Dos picos: 4 meses y 8 años.
- Afecta más la cara, palmas y plantas, y puede tener lesiones generalizadas.
- Puede tener lesiones localizadas y afectación genital.
- Penfigoide ampolloso inducido por fármacos:
- Cuadro clínico similar al cuadro clásico, pero a edades más tempranas (adultos jóvenes) y, en promedio, 3 meses después de la introducción del fármaco.
- Control rápido después de la suspension del medicamento.
- Penfigoide ampolloso localizado
- Lesiones en un área específica del cuerpo.
- Asociación con radiación, procedimiento quirúrgico y trasplante.
- Liquen plano penfigoide
- Pápulas y placas violáceas pruriginosas, parecidas al liquen plano, en las extremidades. Luego aparecen vesículas y ampollas debajo de las lesiones liquenoides y sobre la piel normal.
- La mucosa oral puede presentar lesiones blancas reticuladas o en encaje.
- Eritrodermia exfoliativa
- Eritema generalizado y descamación incluso en ausencia de ampollas.
- Otras variantes clínicas
- Penfigoide dishidrisiforme.
- Penfigoide gestacional.
Factores de riesgo:
- Edad avanzada.
- HLA DQB1*03:01: Relación con penfigoide ampolloso en caucásicos y también con enfermedades neurológicas.
- Enfermedades neurológicas:
- Esclerosis múltiple, demencia, enfermedad de Alzheimer, enfermedad de Parkinson, epilepsia, esquizofrenia, apoplejía.
- Precede al penfigoide ampolloso en 5.5 años.
- Esclerosis múltiple que tiene la asociación más alta (5-12 veces mayor riesgo).
- Enfermedades autoinmunes:
- Psoriasis, artritis reumatoide, lupus eritematoso, liquen plano, nefropatía membranosa, anemia perniciosa, cirrosis biliar primaria, tiroiditis, polimiositis.
- El penfigoide ampolloso puede aparecer sobre lesiones previas de enfermedades de la piel.
- Neoplasias:
- Controvertido, puede estar relacionado solo con la edad de aparición del penfigoide ampolloso.
- Neoplasias hematológicas: Linfoma de Hodgkin, linfoma no folicular, linfoma de células T y NK maduras, linfoma no Hodgkin, leucemia mieloide, leucemia linfoide, etc.
- Neoplasias no hematológicas: Cáncer gástrico, colorrectal, renal, de vejiga, de próstata, de laringe, de pulmón y de mama.
- Enfermedad cardiovascular:
- Tromboembolismo, ACV, tromboembolismo venoso, embolismo pulmonar.
Factores inductores o exacerbantes:
- Medicamentos:
- Inhibidores de la dipeptidil-peptidasa IV o DPP-IV (sitagliptina, vildagliptina y linagliptina, etc), diuréticos (furosemida y espironolactona), antiinflamatorios no esteroideos (diclofenaco, ibuprofeno, etc), ácido acetilsalicílico, antibióticos (amoxicilina, ampicilina, cefalexina, ciprofloxacina, levofloxacina, metronidazol, penicilina, rifampicina, etc), griseofulvina, amlodipina, nifedipina, captopril, enalapril, atenolol, losartán, antipsicóticos, yoduro de potasio, anti-TNF alfa (adalimumab, etanercept, infliximab, efalizumab), anti -PD1 y PDL-1 (Pembrolizumab, Nivolumab y Durvalumab), Benzoato de Bencilo, 5-fluorouracilo, etc.
- Vacunas:
- Controversial.
- Penfigoide ampolloso en niños y adolescentes.
- Fototerapia.
- Radioterapia.
- Terapia fotodinámica.
- Radiación ultravioleta (UVA y UVB).
- Quemaduras eléctricas y térmicas.
- Procedimientos quirúrgicos.
- Trauma.
- Trasplante (raro).
- Infección:
- Herpes virus, citomegalovirus, Epstein-Barr, HHV-6, HHV-7, herpes zoster, VIH, hepatitis B, hepatitis C, Toxoplasma gondii, Helicobacter pylori, Sarcoptes scabiei, etc.
Enfoque diagnóstico
El diagnóstico se basa en las características clínicas, la histopatología, la inmunofluorescencia y la serología.
Histopatología:
- Realizado con biopsia de la ampolla integra.
- Penfigoide no ampolloso: Espongiosis eosinofílica e infiltrado inflamatorio dérmico mixto, predominantemente eosinofílico.
- Penfigoide ampolloso clásico: Desprendimiento subepidérmico con eosinófilos (predominante), neutrófilos y fibrina en el contenido de la ampolla e infiltrado inflamatorio dérmico eosinofílico.
Inmunofluorescencia directa:
- Realizado con biopsia de piel perilesional.
- Gold estándar en penfigoide ampolloso.
- Depósito lineal de IgG y/o C3 en la zona de la membrana basal.
Inmunofluorescencia indirecta:
- Realizado bajo piel normal y con sangre del paciente.
- Depósito lineal de IgG en la unión dermoepidérmica.
- Técnica salt split: Depósito de IgG en la zona epidérmica de separación, es decir, en el techo de la ampolla (diferente a otras dermatosis ampollosas subepidérmicas).
Cuantificación de autoanticuerpos circulantes frente a BP180 y BP230 (ELISA):
- No está indicado como test único o primera prueba para el diagnóstico de penfigoide buloso.
- Correlación de la actividad de la enfermedad con los niveles séricos.
- Puede ser utilizado para el seguimiento del tratamiento, especialmente en el momento de su suspensión.
Otros exámenes:
- Hemograma: 50% de los casos presentan neutrofilia o eosinofilia.
- Aumento de IgE: Ayuda a determinar los pacientes que se beneficiarán del uso de omalizumab.
- Investigación de neoplasias, según se sospeche.
- Bioquímica, serologías (para la posible programación de tratamiento con inmunosupresores).
Diagnóstico diferencial
- Penfigoide cicatricial o de las membranas mucosas.
- Epidermólisis ampollosa adquirida.
- Pénfigo foliáceo.
- Pénfigo vulgar.
- Pénfigo herpetiforme.
- Dermatosis ampollosa por IgA lineal.
- Lupus eritematoso ampolloso.
- Dermatitis atópica.
- Dermatitis de contacto.
- Dermatitis herpetiforme.
- Urticaria.
- Prurigo.
- Impétigo.
- Eritema multiforme.
- Necrolisis epidérmica toxica.
- Síndrome de Sweet.
- Eritema pigmentario fijo.
Seguimiento
Puede ser autolimitado, pero puede tardar meses o años en resolverse.
Las complicaciones que conducen a la hospitalización (infección secundaria, sepsis, neumonía, infección del tracto urinario, eritrodermia) y los efectos adversos de los medicamentos aumentan el riesgo de mortalidad entre 3 y 6 veces.
La edad avanzada, el trastorno neurológico, el aumento del nivel sérico de IgG anti-BP180 tienen un mayor riesgo de mortalidad. Los ahorradores de corticosteroides reducen la tasa de mortalidad.
El penfigoide ampolloso inducido por fármacos tiene un mejor pronóstico con una buena respuesta al tratamiento después de retirar el medicamento causante, con poca o ninguna recurrencia.
El penfigoide ampolloso en niños casi siempre logra la remisión con terapia con corticosteroides y medicación adyuvante.
Complicaciones
Aumento del riesgo de trombosis, con un riesgo de mortalidad del 23.5% a 1 año.
Enfoque terapéutico
El tratamiento ayuda a prevenir la aparición de nuevas lesiones, curar las existentes y controlar el prurito.
Cuidado general:
- Lavar las ampollas con jabón antiséptico.
- Las áreas ulceradas deben cubrirse con apósitos no adherentes.
Enfermedad leve a moderada o localizada (< 5-10 ampollas nuevas por día o lesiones en un solo sitio):
- Corticosteroide tópico de alta potencia (primera línea de tratamiento): Propionato de clobetasol.
- Puede requerir corticosteroides orales + adyuvante ahorrador de corticosteroides (dapsona o doxiciclina con o sin nicotinamida).
Enfermedad moderada a grave o extensa (>10 ampollas nuevas por día):
- Corticosteroide oral (1.ª línea de tratamiento) + adyuvante ahorrador de corticosteroides (Metotrexato, Azatioprina o Micofenolato mofetilo).
- Idealmente, el adyuvante ahorrador de corticosteroides debe iniciarse desde el inicio del corticosteroide o durante su destete para minimizar los efectos adversos de la terapia crónica con corticosteroides.
- Si existe una contraindicación para los corticosteroides sistémicos, solo se deben usar medicamentos ahorradores de corticosteroides.
Casos graves que no mejoran con la corticoterapia sistémica (resistentes o recalcitrantes o con progresión rápida):
- Metilprednisolona, Inmunoglobulina, Rituximab, Dupilumab u Omalizumab (este último para casos de IgE sérica elevada y/o eosinofilia).
Prescripción ambulatoria
Enfermedad leve a moderada o localizada
Recomendaciones:
- Definición: < 5-10 ampollas nuevas por día o lesiones en un solo sitio.
- El tratamiento ayuda a prevenir la aparición de nuevas lesiones, curar las existentes y controlar el prurito.
- Idealmente, el adyuvante ahorrador de corticosteroides debe iniciarse desde el inicio del corticosteroide o durante su destete para minimizar los efectos adversos de la terapia crónica con corticosteroides.
- Si existe una contraindicación para los corticosteroides sistémicos, solo se deben usar medicamentos ahorradores de corticosteroides.
Cuidado general:
- Lavar las ampollas con jabón antiséptico.
- Las áreas ulceradas deben cubrirse con apósitos no adherentes.
Tratamiento farmacológico
Elija uno de los esquemas a continuación según sea necesario clínicamente.
Esquema A: Corticoide tópico de alta potencia (primera línea de tratamiento):
- Propionato de clobetasol (crema o pomada al 0.05%): Aplicar sobre las lesiones, excepto la cara, 1-2 veces al día durante 4-12 meses (dosis máxima: 30 g/día):
- Reducir la dosis gradualmente después de 15 días o después del control clínico (sin nuevas lesiones cutáneas).
- Sugerencia de destete: 10-20 g 1 vez al día en el primer mes; días alternos en el segundo mes; 2 veces por semana en el tercer mes; 1 vez por semana en el cuarto mes; 10 g 1 vez por semana en áreas previamente afectadas por otros 8 meses.
Esquema B: Si las lesiones persisten: Asociación:
- Prednisona 0.5 – 0.75 mg/kg/día VO 1 vez al día con destete gradual a los 15 días o tras control clínico. +
- Adyuvante ahorrador de corticoides.
Recomendaciones para la prednisona:
- Sugerencia de destete: Reducir 0.1 mg/kg/día durante 4-6 meses y tratamiento total durante 9-12 meses.
- En caso de recaída, volver al tratamiento inicial.
Opciones de adyuvantes ahorradores de corticoides:
- Dapsona (100 mg/comprimido) 1 comprimido VO 1 vez al día o, 0.5-2 mg/kg/día VO 1 vez al día en niños hasta una mejoría clínica completa y sostenida:
- Especialmente indicado cuando existe lesión de mucosas.
- Doxiciclina (100 mg/comprimido) 1 comprimido por vía oral cada 12 horas hasta una mejoría clínica completa y sostenida:
- Usar solo en > 12 años de edad y puede combinarse con nicotinamida.
- Nicotinamida (500 mg/comprimido) 1 o 2 comprimidos VO 1-2 veces al día hasta una mejoría clínica completa y mantenida:
- Usar solo en asociación con doxiciclina.
Enfermedad moderada a grave o extensa
Recomendaciones:
- Definición: > 10 ampollas nuevas por día.
- El tratamiento ayuda a prevenir la aparición de nuevas lesiones, curar las existentes y controlar el prurito.
- Idealmente, el adyuvante ahorrador de corticosteroides debe iniciarse desde el inicio del corticosteroide o durante su destete para minimizar los efectos adversos de la terapia crónica con corticosteroides.
- Si existe una contraindicación para los corticosteroides sistémicos, solo se deben usar medicamentos ahorradores de corticosteroides.
Cuidado general:
- Lavar las ampollas con jabón antiséptico.
- Las áreas ulceradas deben cubrirse con apósitos no adherentes.
Tratamiento farmacológico
Enfermedad moderada a grave o extensa: Asociación:
- Prednisona 0.5-1 mg/kg/día VO 1 vez al día con destete gradual a los 15 días o tras control clínico. +
- Adyuvante ahorrador de corticoides.
Recomendaciones para la prednisona:
- Sugerencia de destete: Reducir 0.1 mg/kg/día durante 4-6 meses y tratamiento total durante 9-12 meses.
- En caso de recaída, volver al tratamiento inicial.
Opciones de adyuvantes ahorradores de corticoides:
- Metotrexato 15 mg VO 1 vez por semana hasta una mejoría clínica completa y sostenida.
- Azatioprina 0.5-2 mg/kg/día VO 1-2 veces al día hasta una mejoría clínica completa y sostenida. Riesgo de hepatotoxicidad.
- Micofenolato mofetilo (500 mg/comprimido) 35-45 mg/kg/día VO 2 veces al día hasta mejoría clínica completa y sostenida (máximo 3 g/día).
Prescripción hospitalaria
Enfermedad resistente o que progresa rápidamente
- Definición: Casos graves que no mejoran con corticoterapia sistémica (resistentes o recalcitrantes o de rápida progresión).
Tratamiento farmacológico
Casos resistentes o rápidamente progresivos: Monoterapia. Elige una de las siguientes opciones:
- Metilprednisolona 1g IV 1 vez al día durante 3-5 días consecutivos. Buena opción para controlar la enfermedad más rápido.
- Inmunoglobulina 2 g/kg/dosis IV total. Aplicar 1 vez al día y la dosis debe estar dividida en 3-5 días consecutivos cada mes y hasta control clínico.
- Rituximab 1 g IV el primer dia y el dia 14, o 375 mg/m² de superficie corporal/semana IV 1 vez por semana durante 4 semanas. Indicado principalmente en casos persistentes o recurrentes.
- Dupilumab: Dosis inicial de 600 mg SC; seguido de 300 mg SC 1 vez cada 2 semanas (14 días).
- Omalizumab 300 mg SC cada 2-4 semanas. Opción en casos severos o recalcitrantes con aumento del nivel de IgE sérica y/o eosinofilia.
(Ver – Ectima)
Referencias bibliográficas
Santi CG, et al. Consensus on the treatment of autoimmune bullous dermatoses: bullous pemphigoid, mucous membrane pemphigoid and epidermolysis bullosa acquisita – Brazilian Society of Dermatology. An Bras Dermatol. 2019; 94 (2 Suppl 1):33-47.
Miyamoto D, et al. Bullous pemphigoid. An Bras Dermatol. 2019; 94(2):133-146.
Pratasava V, et al. Bullous Pemphigoid and Other Pemphigoid Dermatoses. Medicina (Kaunas). 2021; 57(10):1061.
Moro F, et al. Bullous Pemphigoid: Trigger and Predisposing Factors. Biomolecules. 2020; 10(10):1432.
Sugerencias y comentarios al correo: contacto@galenbook.com